Сисакян А. С. — Эпидемиологические аспекты идиопатической дилатационной кардиомиопатии
ИДИОПАТИЧЕСКОЙ ДИЛАТАЦИОННОЙ КАРДИОМИОПАТИИ
Сисакян А. С., Гуревич М.А.
Ереванский ГМУ, Медицинский центр Эребуни, отделение сердечно-сосудистой диагностики; МОНИКИ им. М.Ф. Владимирского, кафедра внутренних болезней №1, Москва
Ключевые слова: идиопатическая дилатационная кардиомиопатия, хроническая сердечная недостаточность, эпидемиология.
Идиопатическая дилатационная кардиомиопатия (ИДКМП) считается одним из труднодиагностируе-мых заболеваний миокарда неизвестной этиологии. По данным комитета экспертов ВОЗ [13], ИДКМП характеризуется расширением полостей сердца и прогрессирующей сердечной недостаточностью. ИДКМП является самой распространенной формой кардиомиопатий и имеет наибольшую клиническую значимость.
Несмотря на значительный прогресс в изучении ИДКМП с использованием современных клинических и морфологических методов исследования, диагностика заболевания зачастую представляет затруднения. Последние связаны с отсутствием во многих клиниках современных методов диагностики, позволяющих дифференцировать ИДКМП от других, более часто встречающихся сердечно-сосудистых патологий, имеющих сходную клиническую симптоматику.
Изучение эпидемиологии ИДКМП, внедрение новых параклинических методов исследования, а также улучшение информированности врачей о кар-диомиопатиях, способствуют облегчению диагностики этого заболевания.
Поданным Фремингемского исследования 1971 г., больные ИДКМП составляли 5% от всех больных с хронической сердечной недостаточностью (ХСН). К 1993 г. процент больных с ИДКМП в группе с ХСН увеличился на 4% [8 ]. В исследовании БОЬУО больные ИДКМП составляли 12,5% от всех больных с ХСН [10]. Заболеваемость ИДКМП в общей популяции , по данным разных авторов, колеблется: так, в России , по данным М.С. Кушаковского, распространенность ИДКМП в общей популяции составляет 7-8 случаев в год на 100 000 населения [2]; шведские исследователи приводят интенсивный показатель заболеваемости ИДКМП, равный 5; соотношение между мужчинами и женщинами — 3:1; средний возраст заболевших — 47 лет [11]. Следует отметить, что, хотя в клинике всем больным проводилась эндомиокарди-альная биопсия для исключения активного миокардита, коронарография выполнялась лишь в отдельных случаях— по усмотрению врача.
Результаты проспективных клинико-патолого-анатомических исследований, включавших 250-тысячное население г.Триеста (Италия), позволили определить показатель заболеваемости ИДКМП, который составил 6,95 случаев в год на 100000 населения [12]. С учётом значительной частоты госпитализаций и производимых вскрытий, а также низкого уровня миграции среди населения города, эти данные, вероятно, наиболее близки к истине. В отличие от некоторых других исследователей, S. Raker et al. не считали критерием исключения дигноза ИДКМП употребление алкоголя и умеренную артериальную гипертонию без гипертрофии миокарда [12].
В последние годы достигнут ощутимый прогресс в изучении этиологии и патогенеза ИДКМП. Показано, что заболевание ИДКМП в ряде случаев является гетерогенным, у 20-30% пациентов выявлена семейная форма. Патогенетическое значение имеют два гена [4, 9]. Первый из них кодирует структурные белки типа дистрофина, входящего в состав глико-протеинового комплекса. Обнаружена также мутация цитоскелетного гена MLP , который кодирует миокардиальный белок LJM. По данным польских авторов, которые изучали семейные случаи ИДКМП в период 1992-1999 гг., было выявлено, что 22% случаев от общей популяции больных ИДКМП являются семейными [3].
Исследователи лаборатории генетики Парижского института кардиологии, изучая распространённость ИДКМП в Европе, также отметили, что, несмотря на неясность этиологии ДКМП, в 20-30 % случаев заболевание имеет семейный характер и наследуется по аутосомно-доминантному типу [5]. В связи с этим, согласно рекомендациям ВОЗ по изучению ДКМП [13], для обозначения поражений миокарда с генетически доказанными мутациями ци-тоскелетных генов следует использовать термин «генетическая ДКМП» [7]. В предыдущей номенклатуре их относили к ИДКМП.
Анализ эпидемиологической ситуации распространённости ИДКМП указывает на определённые трудности ее оценки [1, 11]. Данное обстоятельство
Российский кардиологический журнал № 4 (42) / 2003
может быть связано с трудностями дифференциальной диагностики ИДКМП и других заболеваний, протекающих с сердечной недостаточностью, при которых также наблюдается дилатация полостей сердца (миокардиты, ишемическая кардиомиопа-тия, алкогольная кардиомиопатия). В ряде случаев указанные заболевания имеют сходную клиническую картину и могут быть отнесены к ИДКМП.
Важность выделения больных ИДКМП из общего числа больных с сердечной недостаточностью в клинической практике обусловлена ещё и тем, что
1. Гуревич М. А. Хроническая сердечная недостаточность, М. 2000г., с. 183.
2. Кушаковский М. С. Хроническая застойная сердечная недостаточность. Идиопатические кардиомиопатии. Санкт-Петербург, 1997.
3. Bilinska Z. T., Michalak E., et al. Phenotypes of familial dilated cardiomyopathy. Eur. Heart J., Aug/Sept.2000, Supply, Vol. 21, p. 147.
4. Ducand J. B., Bachinski L.L.. Localization of a gene responsible for familial dilated cardiomyopathy to chromosome 1q 32. Circulation 1995, Dec. 15, 92 (12), p. 3387-9.
5. Frederique Tesson, Nicolas Sylvius. Epidemiology of cardiac action and desmin gene mutation in a European population of dilated cardiomyopathy. Eur. Heart J., Aug/Sept.2000, Suppl, Vol. 21, p. 148.
6. Kriett J. M., Kaye M. P.. The Registry of the International Society for Heart Transplantation. Eight Official Report, 1998, J. Heart Transplantation, 1998, Vol. 9, p. 323.
7. Li D., Tapscoft T., еt al. Desmin mutation responsible for idiopathic dilated cardiomyopathy. Circulation, 1999, Aug. 3, 100 (5), p. 461-464.
эти больные составляют основной контингент, нуждающийся в трансплантации сердца.Так, по данным .Т. М.КйеН , в США из 3500 тысяч ежегодно проводимых операций по трансплантации сердца 50% выполнены больным ИДКМП [6].
Изучению ИДКМП способствует совершенствование диагностических методов исследования, улучшение оснащённости специализированных клиник и осуществление поэтапного и квалифицированного обследования больных с сердечной недостаточностью.
8. Mckee P. A., Castelli W. P., et al. The natural history of congestive heart failure. The Framingham Study. N. Engl. J. Med., 1971, Vol. 285, p. 1441.
9. Milasin J., Muntoni F., et al. Evidence for dystrophin gene 5 and involvement in X- linked dilated cardiomyopathy. Eur.Heart J. 1999, Aug/Sept., Supply, Vol. 20, p. 165.
10. SOLVD Investigation. Effect of enalapril on survival in patients with reduced left ventricular ejection fraction and congestive heart failure. N. Engl. J. Med.,1991, Vol. 325, p. 293-302.
11. Tearlink J., Goldhaber S. Z., Preiffer M. A.. An overview of contemporary etiologies of congestive heart failure. Am. Heart J., 1991, Vol. 121, N6, p. 1852-1853.
12. Raker S., Sinagra G., Dilenarda A. et al. Epidemiology of dilated cardiomyopathy. A prospective post-mortem study of 5252 necropsies. The Heart Muscle Disease Study Group. Eur. Heart J.-1997-Vol. 18, p. 117-123.
13. Report of 1997,WHO, on the definition and classification of car-diomyopathy.
Определение кардиомиопатии (КМП) в настоящее время носит самый общий характер — это заболевание миокарда, приводящее к дисфункции сердца. Однако характерной чертой для всех вариантов болезни является формирование структурных изменений в миокарде и развитие недостаточности кровообращения. Попытки систематизировать данные аномалии основаны на преобладающих патофизиологических, этиологических или патогенетических факторах.

Отдельный раздел составляют так называемые неклассифицированные кардиомиопатии, к которым относят некомпактный миокард, фиброэластоз, митохондриальные заболевания и др. Кроме того, существуют специфические кардиомиопатии — патология миокарда, сочетающаяся с другими сердечными или системными заболеваниями. Основанием для диагноза кардиомиопатия в этих случаях является несоответствие тяжести нарушения функции сердца и степени первичного заболевания (коронарной или клапанной патологии, артериальной гипертензии). Сюда же относят поражения сердца метаболического происхождения (эндокринные, связанные с дефицитом некоторых субстанций, патологическими инфильтрациями миокарда, амилоидной болезнью), сопряженные с системными заболеваниями соединительной ткани, мышечными дистрофиями, аллергическими и токсическими реакциями, и некоторые другие.
Точная частота заболеваний неизвестна, так как во многих случаях они протекают бессимптомно. Выявляемость первичных КМП в детском возрасте колеблется в широких пределах. Так, в Финляндии на 100 000 детей она составляет 0,65, в США — 1,13, в Австралии — 1,24. Основное количество (более 50%) случаев представлено дилатационной кардиомиопатией (ДКМП), около 40% — гипертрофической кардиомиопатией (ГКМП) и остальные — неспецифическими кардиопатиями. Частота заболевания у грудных детей почти в 12 раз выше, чем в остальных возрастных группах. Среди плодов с патологией сердца КМП составляют 8,9% случаев, среди новорожденных — 3%.
Естественное течение кардиомиопатий. Кардиомиопатии представляют серьезную проблему у детей: около 40% из них умирают или требуют трансплантации сердца в течение двух лет после появления клинических симптомов. При выявлении патологии во внутриутробном периоде около 13% беременностей прерывают, 63% плодов погибает в перинатальном периоде.
Наиболее часто встречающиеся кардиомиопатии у новорожденных представлены ниже.
— Вернуться в оглавление раздела «Кардиология.»
Читайте также:
- Актиномикоз (патогенез, клиника, диагностика, лечение).
- Актиномикоз в области головы. Этиология, патогенез, лечение, профилактика
- Аритмии. Определение, этиология, патогенез, клиника, диагностика, лечение, профилактика.
- Артериальная гиперемия: виды, причины, механизмы развития, внешние признаки и их патогенез. Исходы (физиологическое и патологическое значение).
- Асфиксия. Этиология, патогенез, стадии развития.
- Ахалазия кардии. Этиология, патогенез, диагностика.
- Брюшной тиф. Эпидемиология. Патогенез. Патоморфология
- В12 дефицитная анемия. Этиология, патогенез, клиника, диагностика, лечение.
- Водный баланс. Виды нарушений водного баланса. Этиология, патогенез и проявления гипер- и дегидратаций.
- Водный баланс. Виды нарушений водного баланса. Этиология, патогенез и проявления гипер- и дегидратаций.
- Врожденные и травматические артериальные и артериовенозные аневризмы. Этиология и патогенез. Клиника, диагностика, оперативное лечение.
- Геморрой: анатомия, патогенез, клиника, осложнения, показания к оперативному лечению.
Классификация кардиомиопатий. Этиология дилатационной кардиомиопатии (ДКМП)
Кардиомиопатии – первичные изолированные поражения миокарда невоспалительного характера неизвестной этиологии (идиопатические), они не имеют связи с клапанными пороками или внутрисердечными шунтами, артериальной или легочной гипертензией, ишемической болезнью сердца или системными заболеваниями (как то: коллагенозы, амилоидоз, гемохроматоз и др.), причем в финальной стадии болезни развиваются тяжелая застойная сердечная недостаточность и сложные нарушения сердечного ритма и проходимости.
Классификация кардиомиопатий следующая:
1) дилатационная кардиомиопатия:
г) при коллагенозах;
4) аритмическая дисплазия правого желудочка;
5) сочетание одного из 4 видов кардиомиопатий с артериальной гипертензией.
Заболевания и факторы, предшествовавшие развитию ДКМП, описаны в приведенной ниже таблице (см. табл.).
Заболевания и факторы, предшествовавшие развитию ДКМП
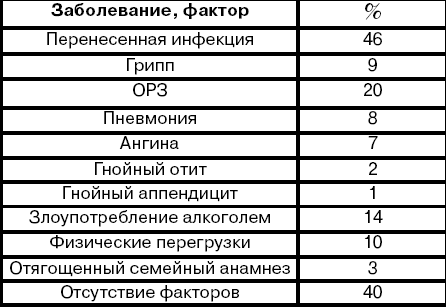
Это самая распространенная форма поражения сердечной мышцы. Заболеваемость составляет 5–8 случаев на 100 000 человек в год. Точного семейного анамнеза у этих пациентов не прослеживается. Мужчины болеют в 2–3 раза чаще женщин.
Патогенез. В результате воспалительного процесса в сердечной мышце (миокардита) происходит гибель отдельных клеток в различных ее участках. Воспаление при этом носит вирусный характер, а клетки, пораженные вирусом, становятся чужеродными агентами для организма. Соответственно, при появлении в организме антигенов развивается комплекс реакций иммунного ответа, направленных на их уничтожение. Постепенно происходит замещение погибших мышечных клеток на соединительную ткань, которая не обладает способностью к растяжимости и сократимости, присущей миокарду. В результате потери основных функций миокарда сердце теряет способность функционировать, как насос. В ответ на это (как компенсаторная реакция) камеры сердца расширяются (т. е. происходит их дилатация), а в оставшейся части миокарда происходит утолщение и уплотнение (т. е. развивается его гипертрофия). Для увеличения доставки кислорода органам и тканям организма возникает стойкое учащение сердечного ритма (синусовая тахикардия).
Данная компенсаторная реакция лишь на время улучшает насосную функцию сердца. Однако возможности дилатации и гипертрофии миокарда ограничиваются количеством жизнеспособного миокарда и являются индивидуальными для каждого конкретного случая заболевания. При переходе процесса в стадию декомпенсации развивается хроническая сердечная недостаточность. Однако на этом этапе вступает в действие еще один компенсаторный механизм: ткани организма увеличивают экстракцию кислорода из крови по сравнению со здоровым организмом. Но этот механизм недостаточен, так как снижение насосной функции сердца ведет к уменьшению поступления в органы и ткани кислорода, который является необходимым для их нормальной жизнедеятельности, при этом количество углекислого газа в них увеличивается.
У 2/3 больных в полостях желудочков на поздних стадиях болезни образуются пристеночные тромбы (вследствие снижения насосной функции сердца, а также при неравномерности сокращения миокарда в камерах сердца) с последующим развитием эмболии по малому или большому кругу кровообращения.
Патогистологические и патоморфологические изменения в сердце. Форма сердца становится шаровидной, масса его увеличивается от 500 до 1000 г, в основном за счет левого желудочка. Миокард становится дряблым, тусклым, с заметными белесоватыми прослойками соединительной ткани, имеется характерное чередование гипертрофированных и атрофичных кардиомиоцитов.
Микроскопически выявляется диффузный фиброз, он может сочетаться как с атрофией, так и с гипертрофией кардиомиоцитов, в которых отмечаются значительное увеличение объема ядер, количества митохондрий, гиперплазия аппарата Гольджи, увеличение количества миофибрилл, свободных и связанных с эндоплазматическим ретикулумом рибосом, обилие гранул гликогена.
Нам важно ваше мнение! Был ли полезен опубликованный материал? Да | Нет
